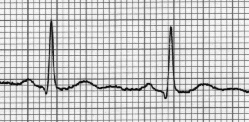
El síndrome de QT corto es una canalopatía hereditaria caracterizada por un anormal acortamiento del intervalo QT (IQT), por un riesgo incrementado para el desarrollo de fibrilación auricular y/o arritmias ventriculares malignas y por la ausencia de cardiopatía estructural. Es una enfermedad heterogénea y se han identificado mutaciones en los genes codificadores de los canales de potasio y de calcio. Un incremento en las corrientes neta de salida de potasio o una disminución en al entrada de calcio favorecen el acortamiento heterogéneo de la repolarización ventricular. La marcada abreviación de la longitud de onda del circuito es un factor arritmogénico adicional. El curso clínico oscila desde formas asintomáticas hasta fibrilación auricular paroxística o permanente, síncope, arritmias ventriculares y muerte súbita. El electrocardiograma muestra IQT 220-360 ms, ondas T altas y puntiagudas, prolongación del intervalo pico-final de la onda T e IQT rígido. Es poco frecuente, pero importante por el riesgo elevado de muerte súbita, que en ocasiones puede ser el debut. Puede presentarse solapado al síndrome de Brugada y a la repolarización precoz. El diagnóstico precisa excluir las causas secundarias que acortan el IQT y la no identificación de una mutación no lo excluye. La estimulación eléctrica programada tiene pobre valor diagnóstico y pronóstico. En los sujetos con muerte súbita abortada o con arritmias ventriculares con compromiso hemodinámica, el desfibrilador es la terapéutica de elección. La quinidina es una opción terapéutica alternativa.
Dra. Marleny Cruz Cardentey

Comentarios recientes